H-index 114, >60,000 citations (Google Scholar)





Publication Highlights

CHOIR improves significant-based detection of cell types and states from single-cell data
Cathrine Sant, Lennart Mucke, M. Ryan Corces
Clustering is a critical step in the analysis of single-cell data, as it enables the discovery and characterization of putative cell types and states. However, most popular clustering tools do not subject clustering results to statistical inference testing, leading to risks of overclustering or underclustering data and often resulting in ineffective identification of cell types with widely differing prevalence. To address these challenges, we present CHOIR (clustering hierarchy optimization by iterative random forests), which applies a framework of random forest classifiers and permutation tests across a hierarchical clustering tree to statistically determine which clusters represent distinct populations. We demonstrate the enhanced performance of CHOIR through extensive benchmarking against 14 existing clustering methods across 100 simulated and 4 real single-cell RNA-seq, ATAC-seq, spatial transcriptomic, and multi-omic datasets. CHOIR can be applied to any single-cell data type and provides a flexible, scalable, and robust solution to the important challenge of identifying biologically relevant cell groupings within heterogeneous single-cell data.
bioRxiv (2024)

Tau ablation in excitatory neurons and postnatal tau knockdown reduce epilepsy, SUDEP, and autism behaviors in a Dravet syndrome model
Eric Shao, Che-Wei Chang, Zhiyong Li, Xinxing Yu, Kaitlyn Ho, Michelle Zhang, Xin Wang, Jeffrey Simms, Iris Lo, Jessica Speckart, Julia Holtzman, Gui-Qiu Yu, Erik D Roberson, Lennart Mucke
Intracellular accumulation of tau aggregates is a hallmark of several neurodegenerative diseases. However, global genetic reduction of tau is beneficial also in models of other brain disorders that lack such tau pathology, suggesting a pathogenic role of nonaggregated tau. Here, conditional ablation of tau in excitatory, but not inhibitory, neurons reduced epilepsy, sudden unexpected death in epilepsy (SUDEP), overactivation of the phosphoinositide 3-kinase–AKT-mammalian target of rapamycin pathway, brain overgrowth (megalencephaly), and autism-like behaviors in a mouse model of Dravet syndrome, a severe epileptic encephalopathy of early childhood. Furthermore, treatment with a tau-lowering antisense oligonucleotide, initiated on postnatal day 10, had similar therapeutic effects in this mouse model. Our findings suggest that excitatory neurons are the critical cell type in which tau has to be reduced to counteract brain dysfunctions associated with Dravet syndrome and that overall cerebral tau reduction could have similar benefits, even when initiated postnatally.
Sci. Transl. Med. 14(642): eabm5527 (2022)
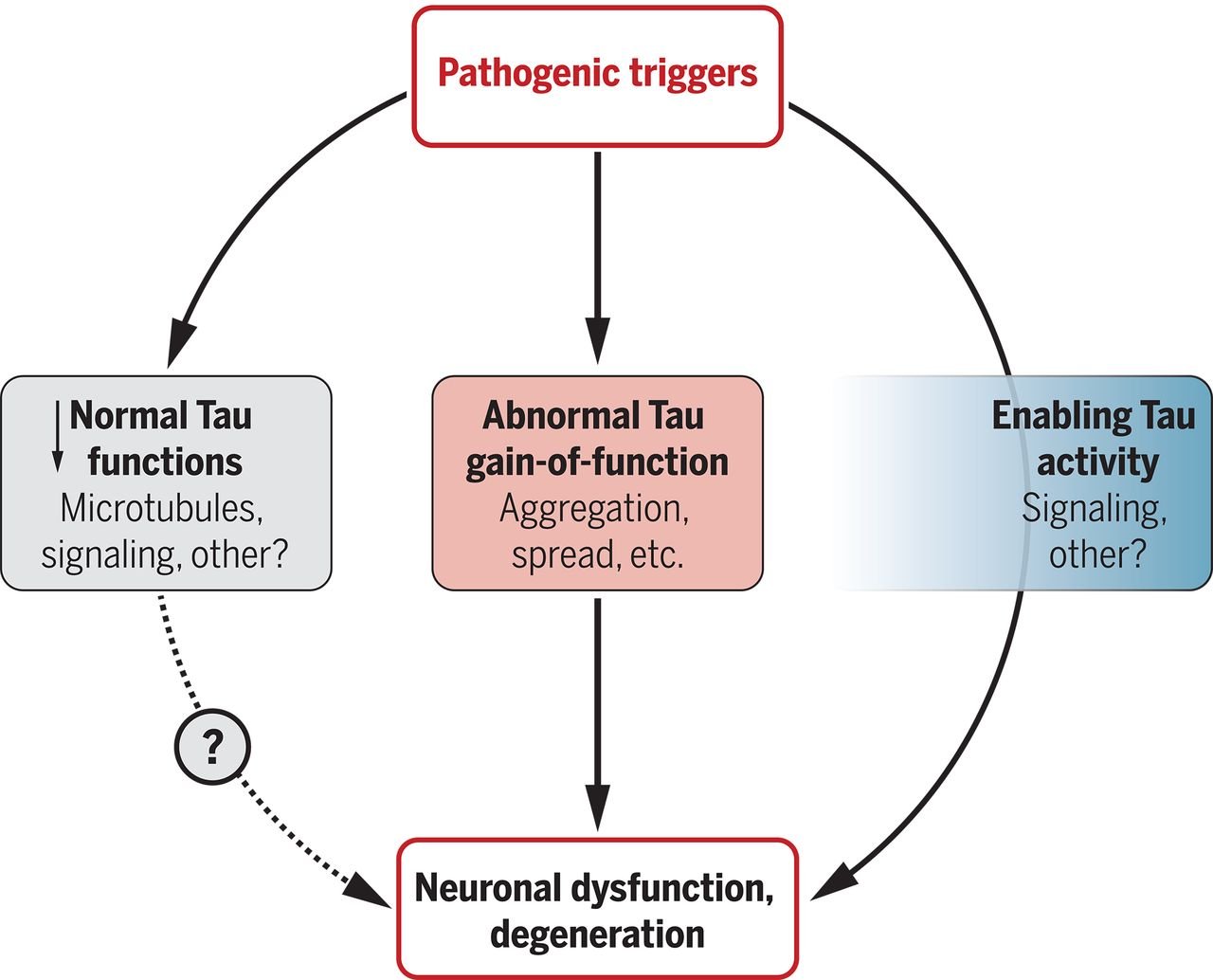
Tau: Enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies
Che-Wei Chang, Eric Shao, Lennart Mucke
The protein tau is implicated in several brain disorders, including Alzheimer's disease, suggesting that it could be a target of therapeutics. However, because it is unclear how the pleiotropic roles of tau lead to neural pathology in different brain diseases, drug development remains challenging. Chang et al. review the possible mechanisms of tau in brain diseases and possible paths forward to improving research and drug development.
Science 371(6532):eabb8255 (2021)

Interdependence of neural network dysfunction and microglial alterations in Alzheimer's disease-related models
Melanie Das, Wenjie Mao, Eric Shao, Soniya Tamhankar, Gui-Qiu Yu, Xinxing Yu, Kaitlyn Ho, Xin Wang, Jiaming Wang, Lennart Mucke
Nonconvulsive epileptiform activity and microglial alterations have been detected in people with Alzheimer’s disease (AD) and related mouse models. However, the relationship between these abnormalities remains to be elucidated. We suppressed epileptiform activity by treatment with the antiepileptic drug levetiracetam or by genetic ablation of tau and found that these interventions reversed or prevented aberrant microglial gene expression in brain tissues of aged human amyloid precursor protein transgenic mice, which simulate several key aspects of AD. The most robustly modulated genes included multiple factors previously implicated in AD pathogenesis, including TREM2, the hypofunction of which increases disease risk. Genetic reduction of TREM2 exacerbated epileptiform activity after mice were injected with kainate. We conclude that AD-related epileptiform activity markedly changes the molecular profile of microglia, inducing both maladaptive and adaptive alterations in their activities. Increased expression of TREM2 seems to support microglial activities that counteract this type of network dysfunction.
iScience 24(11): 103245 (2021)

Tau reduction affects excitatory and inhibitory neurons differently, reduces excitation/inhibition ratios, and counteracts network hypersynchrony
Che-Wei Chang, Mark D Evans, Xinxing Yu, Gui-Qiu Yu, Lennart Mucke
The protein tau has been implicated in many brain disorders. In animal models, tau reduction suppresses epileptogenesis of diverse causes and ameliorates synaptic and behavioral abnormalities in various conditions associated with excessive excitation-inhibition (E/I) ratios. However, the underlying mechanisms are unknown. Global genetic ablation of tau in mice reduces the action potential (AP) firing and E/I ratio of pyramidal cells in acute cortical slices without affecting the excitability of these cells. Tau ablation reduces the excitatory inputs to inhibitory neurons, increases the excitability of these cells, and structurally alters their axon initial segments (AISs). In primary neuronal cultures subjected to prolonged overstimulation, tau ablation diminishes the homeostatic response of AISs in inhibitory neurons, promotes inhibition, and suppresses hypersynchrony. Together, these differential alterations in excitatory and inhibitory neurons help explain how tau reduction prevents network hypersynchrony and counteracts brain disorders causing abnormally increased E/I ratios.
Cell Rep. 37(3):109855 (2021)

Behavioral and neural network abnormalities in human APP transgenic mice resemble those of App knock-in mice and are modulated by familial Alzheimer’s disease mutations but not inhibition of BACE1
Erik CB Johnson, Kaitlyn Ho, Gui-Qiu Yu, Melanie Das, Pascal E Sanchez, Biljana Djukic, Isabel Lopez, Xinxing Yu, Michael Gill, Weiping Zhang, Jeanne T Paz, Jorge J Palop, Lennart Mucke
Alzheimer's disease (AD) is the most frequent and costly neurodegenerative disorder. Although diverse lines of evidence suggest that the amyloid precursor protein (APP) is involved in its causation, the precise mechanisms remain unknown and no treatments are available to prevent or halt the disease. A favorite hypothesis has been that APP contributes to AD pathogenesis through the cerebral accumulation of the amyloid-β peptide (Aβ), which is derived from APP through sequential proteolytic cleavage by BACE1 and γ-secretase. However, inhibitors of these enzymes have failed in clinical trials despite clear evidence for target engagement.
Mol. Neurodegener. 15:53, 1–26 (2020)

Tau reduction prevents key features of autism in mouse models
Chao Tai, Che-Wei Chang, Gui-Qiu Yu, Isabel Lopez, Xinxing Yu, Xin Wang, Weikun Guo, Lennart Mucke
Autism is characterized by repetitive behaviors, impaired social interactions, and communication deficits. It is a prevalent neurodevelopmental disorder, and available treatments offer little benefit. Here, we show that genetically reducing the protein tau prevents behavioral signs of autism in two mouse models simulating distinct causes of this condition. Similar to a proportion of people with autism, both models have epilepsy, abnormally enlarged brains, and overactivation of the phosphatidylinositol 3-kinase (PI3K)/Akt (protein kinase B)/ mammalian target of rapamycin (mTOR) signaling pathway. All of these abnormalities were prevented or markedly diminished by partial or complete genetic removal of tau. We identify disinhibition of phosphatase and tensin homolog deleted on chromosome 10 (PTEN), a negative PI3K regulator that tau controls, as a plausible mechanism and demonstrate that tau interacts with PTEN via tau’s proline-rich domain. Our findings suggest an enabling role of tau in the pathogenesis of autism and identify tau reduction as a potential therapeutic strategy for some of the disorders that cause this condition.
Neuron 106(3):421–437.e112 (2020)

Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease
Niraj M Shanbhag, Mark D Evans, Wenjie Mao, Alissa L Nana, William W Seeley, Anthony Adame, Robert A Rissman, Eliezer Masliah, Lennart Mucke
The maintenance of genomic integrity is essential for normal cellular functions. However, it is difficult to maintain over a lifetime in postmitotic cells such as neurons, in which DNA damage increases with age and is exacerbated by multiple neurological disorders, including Alzheimer’s disease (AD). Here we used immunohistochemical staining to detect DNA double strand breaks (DSBs), the most severe form of DNA damage, in postmortem brain tissues from patients with mild cognitive impairment (MCI) or AD and from cognitively unimpaired controls. Immunostaining for γH2AX—a post-translational histone modification that is widely used as a marker of DSBs—revealed increased proportions of γH2AX-labeled neurons and astrocytes in the hippocampus and frontal cortex of MCI and AD patients, as compared to age-matched controls. In contrast to the focal pattern associated with DSBs, some neurons and glia in humans and mice showed diffuse pan-nuclear patterns of γH2AX immunoreactivity. In mouse brains and primary neuronal cultures, such pan-nuclear γH2AX labeling could be elicited by increasing neuronal activity. To assess whether pan-nuclear γH2AX represents DSBs, we used a recently developed technology, DNA damage in situ ligation followed by proximity ligation assay, to detect close associations between γH2AX sites and free DSB ends. This assay revealed no evidence of DSBs in neurons or astrocytes with prominent pan-nuclear γH2AX labeling. These findings suggest that focal, but not pan-nuclear, increases in γH2AX immunoreactivity are associated with DSBs in brain tissue and that these distinct patterns of γH2AX formation may have different causes and consequences. We conclude that AD is associated with an accumulation of DSBs in vulnerable neuronal and glial cell populations from early stages onward. Because of the severe adverse effects this type of DNA damage can have on gene expression, chromatin stability and cellular functions, DSBs could be an important causal driver of neurodegeneration and cognitive decline in this disease.
Acta Neuropathol. Commun. 7:77, 1–18 (2019)

Klotho controls the brain-immune system interface in the choroid plexus
Lei Zhu, Liana R Stein, Daniel Kim, Kaitlyn Ho, Gui-Qiu Yu, Lihong Zhan, Tobias E Larsson, Lennart Mucke
Located within the brain's ventricles, the choroid plexus produces cerebrospinal fluid and forms an important barrier between the central nervous system and the blood. For unknown reasons, the choroid plexus produces high levels of the protein klotho. Here, we show that these levels naturally decline with aging. Depleting klotho selectively from the choroid plexus via targeted viral vector-induced knockout in Klothoflox/flox mice increased the expression of multiple proinflammatory factors and triggered macrophage infiltration of this structure in young mice, simulating changes in unmanipulated old mice. Wild-type mice infected with the same Cre recombinase-expressing virus did not show such alterations. Experimental depletion of klotho from the choroid plexus enhanced microglial activation in the hippocampus after peripheral injection of mice with lipopolysaccharide. In primary cultures, klotho suppressed thioredoxin-interacting protein-dependent activation of the NLRP3 inflammasome in macrophages by enhancing fibroblast growth factor 23 signaling. We conclude that klotho functions as a gatekeeper at the interface between the brain and immune system in the choroid plexus. Klotho depletion in aging or disease may weaken this barrier and promote immune-mediated neuropathogenesis.
Proc. Natl. Acad. Sci. USA. 115:48, E11388–E11396 (2018)

Istradefylline reduces memory deficits in aging mice with amyloid pathology
Anna G Orr, Iris Lo, Heike Schumacher, Kaitlyn Ho, Michael Gill, Weikun Guo, Daniel H Kim, Anthony Knox, Takashi Saito, Takaomi C Saido, Jeffrey Simms, Carlee Toddes, Xin Wang, Gui-Qiu Yu, Lennart Mucke
Adenosine A2A receptors are putative therapeutic targets for neurological disorders. The adenosine A2A receptor antagonist istradefylline is approved in Japan for Parkinson's disease and is being tested in clinical trials for this condition elsewhere. A2A receptors on neurons and astrocytes may contribute to Alzheimer's disease (AD) by impairing memory. However, it is not known whether istradefylline enhances cognitive function in aging animals with AD-like amyloid plaque pathology. Here, we show that elevated levels of Aβ, C-terminal fragments of the amyloid precursor protein (APP), or amyloid plaques, but not overexpression of APP per se, increase astrocytic A2A receptor levels in the hippocampus and neocortex of aging mice. Moreover, in amyloid plaque-bearing mice, low-dose istradefylline treatment enhanced spatial memory and habituation, supporting the conclusion that, within a well-defined dose range, A2A receptor blockers might help counteract memory problems in patients with Alzheimer's disease.
Neurobiol. Dis. 110: 29–36 (2018)

Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease
Keith A Vossel, Kamalini G Ranasinghe, Alexander J Beagle, Danielle Mizuiri, Susanne M Honma, Anne F Dowling, Sonja M Darwish, Victoria Van Berlo, Deborah E Barnes, Mary Mantle, Anna M Karydas, Giovanni Coppola, Erik D Roberson, Bruce L Miller, Paul A Garcia, Heidi E Kirsch, Lennart Mucke, Srikantan S Nagarajan
Seizures are more frequent in patients with Alzheimer's disease (AD) and can hasten cognitive decline. However, the incidence of subclinical epileptiform activity in AD and its consequences are unknown. Motivated by results from animal studies, we hypothesized higher than expected rates of subclinical epileptiform activity in AD with deleterious effects on cognition.
Ann. Neurol. 80: 858–870 (2016)

Network abnormalities and interneuron dysfunction in Alzheimer disease
Jorge J Palop and Lennart Mucke
The function of neural circuits and networks can be controlled, in part, by modulating the synchrony of their components' activities. Network hypersynchrony and altered oscillatory rhythmic activity may contribute to cognitive abnormalities in Alzheimer disease (AD). In this condition, network activities that support cognition are altered decades before clinical disease onset, and these alterations predict future pathology and brain atrophy. Although the precise causes and pathophysiological consequences of these network alterations remain to be defined, interneuron dysfunction and network abnormalities have emerged as potential mechanisms of cognitive dysfunction in AD and related disorders. Here, we explore the concept that modulating these mechanisms may help to improve brain function in these conditions.
Nat. Rev. Neurosci. 17: 777–792 (2016)

Network dysfunction in α-synuclein transgenic mice and human Lewy body dementia
Meaghan Morris, Pascal E Sanchez, Laure Verret, Alexander J Beagle, Weikun Guo, Dena Dubal, Kamalini G Ranasinghe, Akihiko Koyama, Kaitlyn Ho, Gui‐Qiu Yu, Keith A Vossel, Lennart Mucke
Dementia with Lewy bodies (DLB) belongs to a family of common neurodegenerative diseases called synucleinopathies, which are pathologically characterized by the mislocalization and aggregation of the small, presynaptic protein α-synuclein (SYN). DLB, Parkinson's disease (PD), and Parkinson's disease with dementia (PDD) are closely related synucleinopathies, but are distinguished by differences in the relative onset of motor and cognitive impairments and in the distribution of SYN pathology. While much is known about the motor impairments in PD, very little is known about the mechanisms of cognitive impairment and cortical network dysfunction in DLB and PDD. DLB and PDD patients have dementia with visual hallucinations, attentional fluctuations, and parkinsonism. These symptoms are associated with a prominent slowing of cortical oscillations on electroencephalography (EEG), resulting in a shift in spectral power from higher (alpha, beta, gamma) to lower (delta, theta) frequency bands. However, it remains uncertain whether the neural network and cognitive dysfunction in these conditions are actually caused by SYN.
Ann. Clin. Transl. Neurol. 2(11): 1012–1028 (2015)

Astrocytic adenosine receptor A2A regulates memory
Anna G Orr, Edward C Hsiao, Max M Wang, Kaitlyn Ho, Daniel H Kim, Xin Wang, Weikun Guo, Jing Kang, Gui-Qiu Yu, Anthony Adame, Nino Devidze, Dena B Dubal, Eliezer Masliah, Bruce R Conklin, Lennart Mucke
Astrocytes express a variety of G protein–coupled receptors and might influence cognitive functions, such as learning and memory. However, the roles of astrocytic Gs-coupled receptors in cognitive function are not known. We found that humans with Alzheimer's disease (AD) had increased levels of the Gs-coupled adenosine receptor A2A in astrocytes. Conditional genetic removal of these receptors enhanced long-term memory in young and aging mice and increased the levels of Arc (also known as Arg3.1), an immediate-early gene that is required for long-term memory. Chemogenetic activation of astrocytic Gs-coupled signaling reduced long-term memory in mice without affecting learning. Like humans with AD, aging mice expressing human amyloid precursor protein (hAPP) showed increased levels of astrocytic A2A receptors. Conditional genetic removal of these receptors enhanced memory in aging hAPP mice. Together, these findings establish a regulatory role for astrocytic Gs-coupled receptors in memory and suggest that AD-linked increases in astrocytic A2A receptor levels contribute to memory loss.
Nat. Neurosci. 18:423–434 (2015)

Tau reduction prevents disease in a mouse model of Dravet syndrome
Ania L Gheyara, Ravikumar Ponnusamy, Biljana Djukic, Ryan J Craft, Kaitlyn Ho, Weikun Guo, Mariel M Finucane, Pascal E Sanchez, Lennart Mucke
Reducing levels of the microtubule-associated protein tau has shown promise as a potential treatment strategy for diseases with secondary epileptic features such as Alzheimer disease. We wanted to determine whether tau reduction may also be of benefit in intractable genetic epilepsies.
Ann. Neurol. 76(3): 443–456 (2014)

Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model
Pascal E Sanchez, Lei Zhu, Laure Verret, Keith A Vossel, Anna G Orr, John R Cirrito, Nino Devidze, Kaitlyn Ho, Gui-Qiu Yu, Jorge J Palop, Lennart Mucke
In light of the rising prevalence of Alzheimer’s disease (AD), new strategies to prevent, halt, and reverse this condition are needed urgently. Perturbations of brain network activity are observed in AD patients and in conditions that increase the risk of developing AD, suggesting that aberrant network activity might contribute to AD-related cognitive decline. Human amyloid precursor protein (hAPP) transgenic mice simulate key aspects of AD, including pathologically elevated levels of amyloid-β peptides in brain, aberrant neural network activity, remodeling of hippocampal circuits, synaptic deficits, and behavioral abnormalities. Whether these alterations are linked in a causal chain remains unknown. To explore whether hAPP/amyloid-β–induced aberrant network activity contributes to synaptic and cognitive deficits, we treated hAPP mice with different antiepileptic drugs. Among the drugs tested, only levetiracetam (LEV) effectively reduced abnormal spike activity detected by electroencephalography. Chronic treatment with LEV also reversed hippocampal remodeling, behavioral abnormalities, synaptic dysfunction, and deficits in learning and memory in hAPP mice. Our findings support the hypothesis that aberrant network activity contributes causally to synaptic and cognitive deficits in hAPP mice. LEV might also help ameliorate related abnormalities in people who have or are at risk for AD.
Proc. Natl. Acad. Sci. USA 109(42): E2895–E2903 (2012)

Amyloid-β/Fyn–induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease
Erik D Roberson, Brian Halabisky, Jong W Yoo, Jinghua Yao, Jeannie Chin, Fengrong Yan, Tiffany Wu, Patricia Hamto, Nino Devidze, Gui-Qiu Yu, Jorge J Palop, Jeffrey L Noebels, Lennart Mucke
Alzheimer's disease (AD), the most common neurodegenerative disorder, is a growing public health problem and still lacks effective treatments. Recent evidence suggests that microtubule-associated protein tau may mediate amyloid-β peptide (Aβ) toxicity by modulating the tyrosine kinase Fyn. We showed previously that tau reduction prevents, and Fyn overexpression exacerbates, cognitive deficits in human amyloid precursor protein (hAPP) transgenic mice overexpressing Aβ. However, the mechanisms by which Aβ, tau, and Fyn cooperate in AD-related pathogenesis remain to be fully elucidated. Here we examined the synaptic and network effects of this pathogenic triad. Tau reduction prevented cognitive decline induced by synergistic effects of Aβ and Fyn. Tau reduction also prevented synaptic transmission and plasticity deficits in hAPP mice. Using electroencephalography to examine network effects, we found that tau reduction prevented spontaneous epileptiform activity in multiple lines of hAPP mice. Tau reduction also reduced the severity of spontaneous and chemically induced seizures in mice overexpressing both Aβ and Fyn. To better understand these protective effects, we recorded whole-cell currents in acute hippocampal slices from hAPP mice with and without tau. hAPP mice with tau had increased spontaneous and evoked excitatory currents, reduced inhibitory currents, and NMDA receptor dysfunction. Tau reduction increased inhibitory currents and normalized excitation/inhibition balance and NMDA receptor-mediated currents in hAPP mice. Our results indicate that Aβ, tau, and Fyn jointly impair synaptic and network function and suggest that disrupting the copathogenic relationship between these factors could be of therapeutic benefit.
J. Neurosci. 31(2): 700–711 (2011)

Transsynaptic progression of amyloid-β-induced neuronal dysfunction within the entorhinal-hippocampal network
Julie A Harris, Nino Devidze, Laure Verret, Kaitlyn Ho, Brian Halabisky, Myo T Thwin, Daniel Kim, Patricia Hamto, Iris Lo, Gui-Qiu Yu, Jorge J Palop, Eliezer Masliah, Lennart Mucke
The entorhinal cortex (EC) is one of the earliest affected, most vulnerable brain regions in Alzheimer's disease (AD), which is associated with amyloid-β (Aβ) accumulation in many brain areas. Selective overexpression of mutant amyloid precursor protein (APP) predominantly in layer II/III neurons of the EC caused cognitive and behavioral abnormalities characteristic of mouse models with widespread neuronal APP overexpression, including hyperactivity, disinhibition, and spatial learning and memory deficits. APP/Aβ overexpression in the EC elicited abnormalities in synaptic functions and activity-related molecules in the dentate gyrus and CA1 and epileptiform activity in parietal cortex. Soluble Aβ was observed in the dentate gyrus, and Aβ deposits in the hippocampus were localized to perforant pathway terminal fields. Thus, APP/Aβ expression in EC neurons causes transsynaptic deficits that could initiate the cortical-hippocampal network dysfunction in mouse models and human patients with AD.
Neuron 68: 428–441 (2010)

Cellular source of apolipoprotein E4 determines neuronal susceptibility to excitotoxic injury in transgenic mice
Manuel Buttini, Eliezer Masliah, Gui-Qiu Yu, Jorge J Palop, Shengjun Chang, Aubrey Bernardo, Carol Lin, Tony Wyss-Coray, Yadong Huang, Lennart Mucke
The lipid transport protein apolipoprotein E (apoE) is abundantly expressed in the brain. Its main isoforms in humans are apoE2, apoE3, and apoE4. ApoE4 is the major known genetic risk factor for Alzheimer's disease and also contributes to the pathogenesis of various other neurological conditions. In the central nervous system, apoE is synthesized by glial cells and neurons, but it is unclear whether the cellular source affects its biological activities. To address this issue, we induced excitotoxic injury by systemic kainic acid injection in transgenic Apoe knockout mice expressing human apoE isoforms in astrocytes or neurons. Regardless of its cellular source, apoE3 expression protected neuronal synapses and dendrites against the excitotoxicity seen in apoE-deficient mice. Astrocyte-derived apoE4, which has previously been shown to have detrimental effects in vitro, was as excitoprotective as apoE3 in vivo. In contrast, neuronal expression of apoE4 was not protective and resulted in loss of cortical neurons after excitotoxic challenge, indicating that neuronal apoE4 promotes excitotoxic cell death. Thus, an imbalance between astrocytic (excitoprotective) and neuronal (neurotoxic) apoE4 expression may increase susceptibility to diverse neurological diseases involving excitotoxic mechanisms.
Am. J. Pathol. 177: 563–569 (2010)

Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model
Erik D Roberson, Kimberly Scearce-Levie, Jorge J Palop, Fengrong Yan, Irene H Cheng, Tiffany Wu, Hilary Gerstein, Gui-Qiu Yu, Lennart Mucke
Many potential treatments for Alzheimer's disease target amyloid-beta peptides (Abeta), which are widely presumed to cause the disease. The microtubule-associated protein tau is also involved in the disease, but it is unclear whether treatments aimed at tau could block Abeta-induced cognitive impairments. Here, we found that reducing endogenous tau levels prevented behavioral deficits in transgenic mice expressing human amyloid precursor protein, without altering their high Abeta levels. Tau reduction also protected both transgenic and nontransgenic mice against excitotoxicity. Thus, tau reduction can block Abeta- and excitotoxin-induced neuronal dysfunction and may represent an effective strategy for treating Alzheimer's disease and related conditions.
Science 316(5825):750–754 (2007)

Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease
Jorge J Palop, Jeannie Chin, Erik D Roberson, Jun Wang, Myo T Thwin, Nga Bien-Ly, Jong Yoo, Kaitlyn O Ho, Gui-Qiu Yu, Anatol Kreitzer, Steven Finkbeiner, Jeffrey L Noebels, Lennart Mucke
Neural network dysfunction may play an important role in Alzheimer's disease (AD). Neuronal circuits vulnerable to AD are also affected in human amyloid precursor protein (hAPP) transgenic mice. hAPP mice with high levels of amyloid-beta peptides in the brain develop AD-like abnormalities, including cognitive deficits and depletions of calcium-related proteins in the dentate gyrus, a region critically involved in learning and memory. Here, we report that hAPP mice have spontaneous nonconvulsive seizure activity in cortical and hippocampal networks, which is associated with GABAergic sprouting, enhanced synaptic inhibition, and synaptic plasticity deficits in the dentate gyrus. Many Abeta-induced neuronal alterations could be simulated in nontransgenic mice by excitotoxin challenge and prevented in hAPP mice by blocking overexcitation. Aberrant increases in network excitability and compensatory inhibitory mechanisms in the hippocampus may contribute to Abeta-induced neurological deficits in hAPP mice and, possibly, also in humans with AD.
Neuron 55: 697–711 (2007)

Aggressive amyloidosis in mice expressing human amyloid peptides with the Arctic mutation
Irene H Cheng, Jorge J Palop, Luke A Esposito, Nga Bien-Ly, Fengrong Yan, Lennart Mucke
The Arctic mutation within the amyloid-beta (Abeta) peptide causes Alzheimer disease. In vitro, Arctic-mutant Abeta forms (proto)fibrils more effectively than wild-type Abeta. We generated transgenic mouse lines expressing Arctic-mutant human amyloid precursor proteins (hAPP). Amyloid plaques formed faster and were more extensive in Arctic mice than in hAPP mice expressing wild-type Abeta, even though Arctic mice had lower Abeta(1-42/1-40) ratios. Thus, the Arctic mutation is highly amyloidogenic in vivo.
Nat. Med. (11):1190–1192 (2004)

Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits
Jorge J Palop, Brian Jones, Lisa Kekonius, Jeannie Chin, Gui-Qiu Yu, Jacob Raber, Eliezer Masliah, Lennart Mucke
Transgenic mice expressing human amyloid precursor proteins (hAPP) and amyloid-beta peptides (Abeta) in neurons develop phenotypic alterations resembling Alzheimer's disease (AD). The mechanisms underlying cognitive deficits in AD and hAPP mice are largely unknown. We have identified two molecular alterations that accurately reflect AD-related cognitive impairments. Learning deficits in mice expressing familial AD-mutant hAPP correlated strongly with decreased levels of the calcium-binding protein calbindin-D28k (CB) and the calcium-dependent immediate early gene product c-Fos in granule cells of the dentate gyrus, a brain region critically involved in learning and memory. These molecular alterations were age-dependent and correlated with the relative abundance of Abeta1-42 but not with the amount of Abeta deposited in amyloid plaques. CB reductions in the dentate gyrus primarily reflected a decrease in neuronal CB levels rather than a loss of CB-producing neurons. CB levels were also markedly reduced in granule cells of humans with AD, even though these neurons are relatively resistant to AD-related cell death. Thus, neuronal populations resisting cell death in AD and hAPP mice can still be drastically altered at the molecular level. The tight link between Abeta-induced cognitive deficits and neuronal depletion of CB and c-Fos suggests an involvement of calcium-dependent pathways in AD-related cognitive decline and could facilitate the preclinical evaluation of novel AD treatments.
Proc. Natl. Acad. Sci. U S A 100(16):9572–9577 (2003)

β-Amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease
Eliezer Masliah, Edward Rockenstein, Isaac Veinbergs, Yutaka Sagara, Margaret Mallory, Makoto Hashimoto, Lennart Mucke
Alzheimer's disease and Parkinson's disease are associated with the cerebral accumulation of β-amyloid and α-synuclein, respectively. Some patients have clinical and pathological features of both diseases, raising the possibility of overlapping pathogenetic pathways. We generated transgenic (tg) mice with neuronal expression of human β-amyloid peptides, α-synuclein, or both. The functional and morphological alterations in doubly tg mice resembled the Lewy-body variant of Alzheimer's disease. These mice had severe deficits in learning and memory, developed motor deficits before α-synuclein singly tg mice, and showed prominent age-dependent degeneration of cholinergic neurons and presynaptic terminals. They also had more α-synuclein-immunoreactive neuronal inclusions than α-synuclein singly tg mice. Ultrastructurally, some of these inclusions were fibrillar in doubly tg mice, whereas all inclusions were amorphous in α-synuclein singly tg mice. β-Amyloid peptides promoted aggregation of α-synuclein in a cell-free system and intraneuronal accumulation of α-synuclein in cell culture. β-Amyloid peptides may contribute to the development of Lewy-body diseases by promoting the aggregation of α-synuclein and exacerbating α-synuclein-dependent neuronal pathologies. Therefore, treatments that block the production or accumulation of β-amyloid peptides could benefit a broader spectrum of disorders than previously anticipated.
Proc. Natl. Acad. Sci. USA 98: 12245–12250 (2001)

High-level neuronal expression of Abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation
Lennart Mucke, Eliezer Masliah, Gui-Qiu Yu, Margaret Mallory, Edward M Rockenstein, Gwen Tatsuno, Kang Hu, Dora Kholodenko, Kelly Johnson-Wood, Lisa McConlogue
Amyloid plaques are a neuropathological hallmark of Alzheimer's disease (AD), but their relationship to neurodegeneration and dementia remains controversial. In contrast, there is a good correlation in AD between cognitive decline and loss of synaptophysin-immunoreactive (SYN-IR) presynaptic terminals in specific brain regions. We used expression-matched transgenic mouse lines to compare the effects of different human amyloid protein precursors (hAPP) and their products on plaque formation and SYN-IR presynaptic terminals. Four distinct minigenes were generated encoding wild-type hAPP or hAPP carrying mutations that alter the production of amyloidogenic Abeta peptides. The platelet-derived growth factor beta chain promoter was used to express these constructs in neurons. hAPP mutations associated with familial AD (FAD) increased cerebral Abeta(1-42) levels, whereas an experimental mutation of the beta-secretase cleavage site (671(M-->I)) eliminated production of human Abeta. High levels of Abeta(1-42) resulted in age-dependent formation of amyloid plaques in FAD-mutant hAPP mice but not in expression-matched wild-type hAPP mice. Yet, significant decreases in the density of SYN-IR presynaptic terminals were found in both groups of mice. Across mice from different transgenic lines, the density of SYN-IR presynaptic terminals correlated inversely with Abeta levels but not with hAPP levels or plaque load. We conclude that Abeta is synaptotoxic even in the absence of plaques and that high levels of Abeta(1-42) are insufficient to induce plaque formation in mice expressing wild-type hAPP. Our results support the emerging view that plaque-independent Abeta toxicity plays an important role in the development of synaptic deficits in AD and related conditions.
J. Neurosci. 20(11):4050–4058 (2000)

Alzheimer’s disease: Apolipoprotein E and cognitive performance
Jacob Raber, Derek Wong, Gui-Qiu Yu, Manuel Buttini, Robert W Mahley, Robert E Pitas, Lennart Mucke
Key proteins implicated in the development of Alzheimer's disease are the β-amyloid precursor protein, which gives rise to the β-amyloid peptides that accumulate in the deteriorating brain, and the different isoforms of apolipoprotein E (apoE). The apoE4 variant increases the risk of developing the disease compared with apoE3. We have tested the spatial memory of transgenic mice carrying human forms of these proteins and find that it is impaired in mice with apoE4 but not those with apoE3, even though the levels of β-amyloid in their brains are comparable. The fact that apoE3, but not apoE4, can protect against cognitive deficits induced by β-amyloid may explain why human apoE4 carriers are at greater risk of developing Alzheimer's than apoE3 carriers.
Nature 404: 352–354 (2000)

Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models
Albert Y Hsia, Eliezer Masliah, Lisa McConlogue, Gui-Qiu Yu, Gwen Tatsuno, Kang Hu, Dora Kholodenko, Robert C Malenka, Roger A Nicoll, Lennart Mucke
Autosomal dominant forms of familial Alzheimer's disease (FAD) are associated with increased production of the amyloid beta peptide, Abeta42, which is derived from the amyloid protein precursor (APP). In FAD, as well as in sporadic forms of the illness, Abeta peptides accumulate abnormally in the brain in the form of amyloid plaques. Here, we show that overexpression of FAD(717V-->F)-mutant human APP in neurons of transgenic mice decreases the density of presynaptic terminals and neurons well before these mice develop amyloid plaques. Electrophysiological recordings from the hippocampus revealed prominent deficits in synaptic transmission, which also preceded amyloid deposition by several months. Although in young mice, functional and structural neuronal deficits were of similar magnitude, functional deficits became predominant with advancing age. Increased Abeta production in the context of decreased overall APP expression, achieved by addition of the Swedish FAD mutation to the APP transgene in a second line of mice, further increased synaptic transmission deficits in young APP mice without plaques. These results suggest a neurotoxic effect of Abeta that is independent of plaque formation.
Proc. Natl. Acad. Sci. USA 96: 3228–3233 (1999)

Isoform-specific effects of human apolipoprotein E on brain function revealed in Apoe knockout mice–Increased susceptibility of females
Jacob Raber, Derek Wong, Manuel Buttini, Matthias Orth, Stefano Bellosta, Robert E Pitas, Robert W Mahley, Lennart Mucke
Apolipoprotein E (apoE) mediates the redistribution of lipids among cells and is expressed at highest levels in brain and liver. Human apoE exists in three major isoforms encoded by distinct alleles (epsilon2, epsilon3, and epsilon4). Compared with APOE epsilon2 and epsilon3, APOE epsilon4 increases the risk of cognitive impairments, lowers the age of onset of Alzheimer's disease (AD), and decreases the response to AD treatments. Besides age, inheritance of the APOE epsilon4 allele is the most important known risk factor for the development of sporadic AD, the most common form of this illness. Although numerous hypotheses have been advanced, it remains unclear how APOE epsilon4 might affect cognition and increase AD risk. To assess the effects of distinct human apoE isoforms on the brain, we have used the neuron-specific enolase (NSE) promoter to express human apoE3 or apoE4 at similar levels in neurons of transgenic mice lacking endogenous mouse apoE. Compared with NSE-apoE3 mice and wild-type controls, NSE-apoE4 mice showed impairments in learning a water maze task and in vertical exploratory behavior that increased with age and were seen primarily in females. These findings demonstrate that human apoE isoforms have differential effects on brain function in vivo and that the susceptibility to apoE4-induced deficits is critically influenced by age and gender. These results could be pertinent to cognitive impairments observed in human APOE epsilon4 carriers. NSE-apoE mice and similar models may facilitate the preclinical assessment of treatments for apoE-related cognitive deficits.
Proc. Natl. Acad. Sci. USA 95: 10914–10919 (1998)

Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer’s disease
Tony Wyss-Coray, Eliezer Masliah, Margaret Mallory, Lisa McConlogue, Kelly Johnson-Wood, Carol Lin, Lennart Mucke
Deposition of amyoid-beta peptide in the central nervous system is a hallmark of Alzheimer's disease and a possible cause of neurodegeneration. The factors that initiate or promote deposition of amyloid-beta peptide are not known. The transforming growth factor TGF-beta1 plays a central role in the response of the brain to injury, and increased TGF-beta1 has been found in the central nervous system of patients with Alzheimer's disease. Here we report that TGF-beta1 induces amyloid-beta deposition in cerebral blood vessels and meninges of aged transgenic mice overexpressing this cytokine from astrocytes. Co-expression of TGF-beta1 in transgenic mice overexpressing amyloid-precursor protein, which develop Alzheimer's like pathology, accelerated the deposition of amyloid-beta peptide. More TGF-beta1 messenger RNA was present in post-mortem brain tissue of Alzheimer's patients than in controls, the levels correlating strongly with amyloid-beta deposition in the damaged cerebral blood vessels of patients with cerebral amyloid angiopathy. These results indicate that overexpression of TGF-beta1 may initiate or promote amyloidogenesis in Alzheimer's disease and in experimental models and so may be a risk factor for developing Alzheimer's disease.
Nature 389(6651):603–606 (1997)
-
Sant C, Mucke L, and Corces MR (2024) CHOIR improves significance-based detection of cell types and states from single-cell data. bioRxiv.
Das M, Mao W, Voskobiynyk Y, Necula D, Lew I, Petersen C, Zahn A, Yu G-Q, Yu X, Smith N, Sayed F, Gan L, Paz JT, and Mucke L (2023) Alzheimer risk-increasing TREM2 variant causes aberrant cortical synapse density and promotes network hyperexcitability in mouse models. Neurobiol. Dis. 186: 106263
Shao E, Chang C-W, Li Z, Yu X, Ho K, Zhang M, Wang X, Simms J, Lo I, Speckart J, Holtzman J, Yu G-Q, Roberson ED, and Mucke L (2022) Tau ablation in excitatory neurons and postnatal tau knockdown reduce epilepsy, SUDEP, and autism behaviors in a Dravet syndrome model. Sci. Transl. Med. 14: eabm5527
Chang C-W, Shao E and Mucke L (2021) Tau: enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science 371: eabb8255
Das M, Mao W, Shao E, Tamhankar S, Yu G-Q, Yu X, Ho K, Wang X, Wang J, and Mucke L (2021) Interdependence of neural network dysfunction and microglial alterations in Alzheimer’s disease-related models. iScience 24: 103245
Chang C-W, Evans MD, Yu X, Yu G-Q, and Mucke L (2021) Tau reduction affects excitatory and inhibitory neurons differentially, reduces excitation/inhibition ratios and counteracts network hypersynchrony. Cell Rep. 37: 109855
Chang C-W, Shao E and Mucke L (2021) Tau: enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science 371: eabb8255
Vossel K, Ranasinghe KG, Beagle AJ, La A, Pook KA, Castro M, Mizuiri D, Honma SM, Venkateswaran N, Koestler M, Zhang W, Mucke L, Howell MJ, Possin KL, Kramer JH, Boxer AL, Miller BL, Nagarajan SS, and Kirsch HE (2021) Effect of levetiracetam on cognition in patients with Alzheimer disease with and without epileptiform activity – A randomized clinical trial. JAMA Neurol. 78: 1345–1354
Gulbranson DR, Ho K, Yu G-Q, Yu X, Das M, Shao E, Kim D, Zhang WJ, Choudhary K, Thomas R, and Mucke L (2021) Phenotypic differences between the Alzheimer’s disease-related hAPP-J20 model and heterozygous Zbtb20 knockout mice. eNeuro 8:ENEURO.0089-21.2021
Diaz-Alonso J, Morishita W, Incontro S, Simms J, Holtzman J, Gill M, Mucke L, Malenka RC, and Nicoll RA (2020) Long-term potentiation is independent of the C-tail of the GluA1 AMPA receptor subunit. eLife 9: e58042
Johnson ECB, Ho K, Yu G-Q, Das M, Sanchez PE, Djukic B, Lopez I, Yu X, Gill M, Zhang W, Paz JT, Palop JJ, and Mucke L (2020) Behavioral and neural network abnormalities in human APP transgenic mice resemble those of App knock-in mice and are modulated by familial Alzheimer’s disease mutations but not by inhibition of BACE1. Mol. Neurodegener. 15:53, 1–26
Davis EJ, Broestl L, Abdulai-Saiku S, Worden K, Bonham LW, Miñones-Moyano E, Moreno AJ, Wang D, Chang K, Williams G, Garay BI, Lobach I, Devidze N, Kim D, Anderson-Bergman C, Yu G-Q, White CC, Harris JA, Miller BL, Bennett DA, Arnold AP, De Jager PL, Palop JJ, Panning B, Yokoyama JS, Mucke L, and Dubal DB (2020) A second X chromosome contributes to resilience in a mouse model of Alzheimer’s disease. Sci. Transl. Med. 12: 1–16
Tai C, Chang C-W, Yu G-Q, Lopez I, Yu X, Wang X, Guo W, and Mucke L (2020) Tau reduction prevents key features of autism in mouse models. Neuron 106: 421–437
Shanbhag N, Evans M, Mao W, Nana A, Seeley W, Adame A, Rissman R, Masliah E, and Mucke L(2019) Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol. Commun. 7:77, 1–18
Merlini M, Rafalski VA, Coronado PRE, Gill MT, Ellisman M, Muthukumar G, Subramanian KS, Ryu JK, Syme CA, Davalos D, Seeley WW, Mucke L, Nelson RB, and Akassoglou K (2019) Fibrinogen induces microglia-mediated spine elimination and cognitive impairment in an Alzheimer’s disease model. Neuron 101: 1099–1108
Zhu L, Stein L, Kim D, Ho K, Yu G-Q, Zhan L, Larsson TE, and Mucke L (2018) Klotho controls the brain–immune system interface in the choroid plexus. PNAS 115 (48) E11388–E11396
Ryu JK, Rafalski VA, Meyer-Franke A, Adams RA, Poda SB, Rios Coronado PE, Østergaard Pedersen L, Menon V, Baeten KM, Sikorski SL, Bedard C, Hanspers K, Bardehle S, Mendiola AS, Davalos D, Machado MR, Chan JP, Plastira I, Petersen MA, Pfaff SJ, Ang KK, Hallenbeck KK, Syme C, Hakozaki H, Ellisman MH, Swanson RA, Zamvil SS, Arkin MR, Zorn SH, Pico AR, Mucke L, Freedman SB, Stavenhagen JB, Nelson RB, and Akassoglou K (2018) Fibrin-targeting immunotherapy protects against neuroinflammation and neurodegeneration. Nat. Immunol. 19: 1212–1223
Howe JR, Bear MF, Golshani P, Klann E, Lipton SA, Mucke L, Sahin M, and Silva AJ (2018) The mouse as a model for neuropsychiatric drug development. Curr. Biol. 28: R909–R914
Das M, Maeda S, Hu B, Yu G-Q, Guo W, Lopez I, Yu X, Tai C, Wang X, and Mucke L (2018) Neuronal levels and sequence of tau modulate the power of brain rhythms. Neurobiol. Dis. 117: 181–188
Martinez-Losa M, Tracy TE, Ma K, Verret L, Clemente-Perez A, Khan AS, Cobos I, Ho K, Gan L, and Mucke L, Alvarez-Dolado M, and Palop J (2018) Nav1.1-overexpressing interneuron transplants restore brain rhythms and cognition in a mouse model of Alzheimer’s disease. Neuron 98: 75–89.e5
Orr AG, Lo I, Schumacher H, Ho K, Gill M, Guo W, Kim DH, Knox A, Saito T, Saido TC, Simms J, Toddes C, Wang X, Yu G-Q, and Mucke L (2018) Istradefylline reduces memory deficits in aging mice with amyloid pathology. Neurobiol. Dis. 110: 29–36
Willsey AJ, Morris MT, Wang S, Willsey HR, Sun N, Teerikorpi N, Baum TB, Cagney G, Bender KJ, Desai TA, Srivastava D, Davis GW, Doudna J, Chang E, Sohal V, Lowenstein DH, Li H, Agard D, Keiser MJ, Shoichet B, von Zastrow M, Mucke L, Finkbeiner S, Gan L, Sestan N, Ward ME, Huttenhain R, Nowakowski TJ, Bellen HJ, Frank LM, Khokha MK, Lifton RP, Kampmann M, Ideker T, State MW, Krogan NJ (2018) The Psychiatric Cell Map Initiative: a convergent systems biological approach to illuminating key molecular pathways in neuropsychiatric disorders. Cell 174: 505–520
Tosh JL, Rickman M, Rhymes E, Norona FE, Clayton E, Mucke L, Isaacs AM, Fisher EMC, and Wiseman FK (2017) The integration site of the APP transgene in the J20 mouse model of Alzheimer’s disease. Wellcome Open Res. 2:84
Miyamoto T, Stein L, Thomas R, Djukic B, Taneja P, Knox J, Vossel K, and Mucke L (2017) Phosphorylation of tau at Y18, but not tau-Fyn binding, is required for tau to modulate NMDA receptor-dependent excitotoxicity in primary neuronal culture. Mol. Neurodegener. 12: 41
Vossel KA, Ranasinghe KG, Beagle AJ, Mizuiri D, Honma SM, Dowling AF, Darwish SM, Van Berlo V, Barnes DE, Mantle M, Karydas AM, Coppola G, Roberson ED, Miller BL, Garcia PA, Kirsch HE,Mucke L, and Nagarajan S (2016) Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann. Neurol. 80: 858–870
Maeda S, Djukic B, Taneja P, Yu G-Q, Lo I, Davis A, Craft R, Guo W, Wang X, Kim D, Ponnusamy R, Gill TM, Masliah E, and Mucke L (2016) Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep. 17: 530–551
Palop JJ and Mucke L (2016) Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 17: 777–792
Miyamoto T, Kim D, Knox JA, Johnson E, and Mucke L (2016) Increasing the receptor tyrosine kinase EphB2 prevents amyloid-β-induced depletion of cell-surface glutamate receptors by a mechanism that requires the PDZ-binding motif of EphB2 and neuronal activity. JBC 291: 1719–1734
Suberbielle E, Djukic B, Evans M, Kim DH, Taneja P, Wang X, Finucane M, Knox J, Ho K, Devidze N, Masliah E, and Mucke L (2015) DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 6: 8897
Morris M, Sanchez PE, Verret L, Beagle AJ, Guo W, Dubal D, Ranasinghe KG, Koyama A, Ho, K, Yu G-Q, Vossel KA, and Mucke L (2015) Network dysfunction in a-synuclein transgenic mice and human Lewy body dementia. Ann. Clin. Transl. Neurol. 2: 1012–1028
Morris M, Knudsen GM, Maeda S, Trinidad JC, Ioanoviciu A, Burlingame AL, and Mucke L (2015) Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 18: 1183–1189
Shields LY, Kim H, Zhu L, Haddad D, Berthet A, Pathak D, Lam M, Ponnusamy R, Diaz-Ramirez LG, Gill TM, Sesaki H, Mucke L, and Nakamura K (2015) Dynamin-related protein 1 (Drp1) is required for normal mitochondrial bioenergetic and synaptic function in CA1 hippocampal neurons. Cell Death Dis. 6: e1725
Vossel KA, Xu JC, Fomenko V, Miyamoto T, Suberbielle E, Knox JA, Ho K, Kim DH, Yu G-Q, and Mucke L (2015) Tau reduction prevents Ab-induced axonal transport deficits by blocking activation of GSK3b. JCB 209: 419–433
Dubal DB, Zhu L, Sanchez PE, Worden K, Broestl L, Johnson E, Ho K, Yu G-Q, Kim D, Betourne A, Kuro-o M, Masliah E, Abraham CR, and Mucke L (2015) Life extension factor klotho prevents mortality and enhances cognition in hAPP transgenic mice. J. Neurosci. 35: 2358–2371
Orr AG, Hsiao EC, Wang MM, Ho K, Kim DH, Wang X, Guo W, Kang J, Yu G-Q, Adame A, Devidze N, Dubal DB, Masliah E, Conklin BR, and Mucke L (2015) Astrocytic adenosine receptor A2A and Gs-coupled signaling regulate memory. Nat. Neurosci. 18: 423–434
Cheng JS, Craft R, Yu G-Q, Ho K, Wang X, Mohan G, Mangnitsky S, Ponnusamy R, and Mucke L (2014) Tau reduction diminishes spatial learning and memory deficits after mild repetitive traumatic brain injury in mice. PLoS One 9: e115765
Minami SS, Min S-W, Krabbe G, Wang C, Zhou Y, Asgarov R, Li Y, Martens LH, Elia LP, Ward ME, Mucke L, Farese Jr RV, and Gan L (2014) Progranulin protects against amyloid b deposition and toxicity in Alzheimer’s disease mouse models. Nat. Med. 20: 1157–1164
Gheyara A, Ponnusamy R, Djukic B, Craft RJ, Ho K, Guo W, Finucane M, Sanchez P, and Mucke L (2014) Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann. Neurol. 76: 443–456
Dubal DB, Yokoyama JS, Zhu L, Broestl L, Worden K, Wang D, Sturm VE, Kim D, Klein E, Yu G-Q, Ho K, Eilertson KE, Yu L, Kuro-o M, De Jager PL, Coppola G, Small GW, Bennett DA, Kramer JH, Abraham CR, Miller BL, and Mucke L (2014) Life extension factor klotho enhances cognition. Cell Rep. 7: 1065–1076
Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, Hegde M, Cornes SB, Henry ML, Nelson AB, Seeley WW, Geschwind MD, Gorno-Tempini ML, Shih T, Kirsch HE, Garcia PA; Miller BL, and Mucke L (2013) Seizures and epileptiform activity in the early stages of Alzheimer’s disease. JAMA Neurology 70: 1158–1166
Suberbielle E, Sanchez PE, Kravitz AV, Wang X, Ho K, Eilertson K, Devidze N, Kreitzer AC, and Mucke L (2013) Physiologic brain activity cause DNA double strand breaks in neurons, with exacerbation by amyloid-b. Nat. Neurosci. 16: 613–621
Morris M, Hamto P, Adame A, Devidze N, Masliah E, and Mucke L (2013) Age-appropriate cognition and subtle dopamine-independent motor deficits in aged Tau knockout mice. Neurobiol. Aging 34: 1523–1529
Harris JA, Koyama A, Maeda S, Ho K, Devidze N, Dubal DB, Yu G-Q, Masliah E, and Mucke L (2012) Human P301L-mutant tau expression in mouse entorhinal-hippocampal network causes tau aggregation and presynaptic pathology but no cognitive deficits. PLoS One 7: e45881
Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu G-Q, Palop JJ, andMucke L (2012) Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. PNAS 109: E2895–2903
Verret L, Mann EO, Hang GB, Barth AMI, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, and Palop JJ (2012) Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149: 708–721
Huang Y and Mucke L (2012) Alzheimer mechanisms and therapeutic strategies. Cell 148: 1204–1222
Minami SS, Sun B, Popat K, Kauppinen T, Pleiss M, Zhou Y, Ward ME, Floreancig P, Mucke L, Desai T, and Gan L (2012) Selective targeting of microglia by quantum dots. J. Neuroinflammation 9: 22
Morris M, Maeda S, Vossel K, and Mucke L (2011) The many faces of tau. Neuron 70: 410–426
Morris M, Koyama A, Masliah E, and Mucke L(2011) Tau reduction does not prevent motor deficits in two mouse models of Parkinson’s disease. PLoS One 6: e29257
Cissé M, Sanchez PE, Kim DH, Ho K, Yu G-Q, and Mucke L (2011) Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J. Neurosci. 31: 10427–10431
Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu G-Q, Palop JJ, Noebels JL, and Mucke L (2011) Amyloid-b/Fyn–induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 31: 700–711
Cissé M, Halabisky B, Harris J, Devidze N, Dubal D, Sun B, Orr A, Lotz G, Kim DH, Hamto P, Ho K, Yu G-Q, and Mucke L(2011) Reversing EphB2 receptor depletion rescues cognitive functions in Alzheimer model. Nature 469: 47–52
Harris JA, Devidze N, Verret L, Ho K, Halabisky B, Thwin MT, Kim D, Hamto P, Lo I, Yu G-Q, Palop JJ, Masliah E, and Mucke L(2010) Transsynaptic progression of amyloid-b-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron 68: 428–441
Vossel K, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, and Mucke L (2010) Tau reduction prevents Aβ-induced defects in axonal transport. Science 330: 198
Palop JJ and Mucke L (2010) Synaptic depression and aberrant excitatory network activity in Alzheimer’s disease: two faces of the same coin?NeuroMolecular Med. 12: 48–55
Sanchez-Mejia RO and Mucke L (2010) Phospholipase A2 and arachidonic acid in Alzheimer’s disease. BBA 1801: 784–790
Palop JJ and Mucke L (2010) Amyloid-β–induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat. Neurosci. 13: 812–818
Buttini M, Masliah E, Yu G-Q, Palop JJ, Chang S, Bernardo A, Lin C, Wyss-Coray T, Huang Y, and Mucke L (2010) Cellular source of apolipoprotein E4 determines neuronal susceptibility to excitotoxic injury in transgenic mice. Am. J. Pathol. 177: 563–569
Harris JA, Devidze N, Halabisky B, Lo I, Thwin MT, Yu G-Q, Bredesen DE, Masliah E, and Mucke L(2010) Many neuronal and behavioral impairments in transgenic mouse models of Alzheimer’s disease are independent of caspase cleavage of the amyloid precursor protein. J. Neurosci. 30: 372–381
Sun B, Halabisky B, Zhou Y, Palop JJ, Yu G-Q, Mucke L, and Gan L (2009) Imbalance between GABAergic and glutamatergic transmission impairs adult neurogenesis in an animal model of Alzheimer's disease. Cell Stem Cell 5: 624–633
Palop JJ and Mucke L (2009) Epilepsy and cognitive impairments in Alzheimer’s disease. Arch. Neurol. 66: 435–440
Meilandt WJ, Cisse M, Ho K, Wu T, Esposito LA, Scearce-Levie K, Cheng IH, Yu G-Q, and Mucke L (2009) Neprilysin overexpression inhibits plaque formation but fails to reduce pathogenic Ab oligomers and associated cognitive deficits in human amyloid precursor protein transgenic mice. J. Neurosci. 29: 1977–1986
Cheng JS, Dubal DB, Kim DH, Legleiter J, Cheng IH, Yu G-Q, Tesseur I, Wyss-Coray T, Bonaldo P, and Mucke L (2009) Collagen VI protects neurons against Ab toxicity. Nat. Neurosci. 12: 119–121
Tamgüney G, Giles K, Glidden DV, Lessard P, Wille H, Tremblay P, Groth DF, Yehiely F, Korth C, Moore RC, Tatzelt J, Rubinstein E, Boucheix C, Yang X, Stanley P, Lisanti MP, Dwek RA, Rudd PM, Moskovitz J, Epstein CJ, Cruz TD, Kuziel WA, Maeda N, Sap J, Ashe KH, Carlson GA, Tesseur I, Wyss-Coray T, Mucke L, Weisgraber KH, Mahley RW, Cohen FE, and Prusiner SB (2008) Genes contributing to prion pathogenesis. J. Gen. Virol. 89: 1777–1788
Sanchez-Mejia RO, Newman JW, Toh S, Yu G.-Q, Zhou Y, Halabisky B, Cisse M, Scearce-Levie K, Cheng IH, Gan L, Palop JJ, Bonventre JV, and Mucke L (2008) Phospholipase A2 reduction ameliorates cognitive deficits in mouse model of Alzheimer’s disease. Nat. Neurosci. 11: 1311–1318
Meilandt WJ, Yu G-Q, Chin J, Roberson ED, Palop JJ, Wu T, Scearce-Levie K, and Mucke L (2008) Enkephalin elevations contribute to neuronal and behavioral impairments in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 28: 5007–5017
Scearce-Levie K, Roberson ED, Gerstein H, Cholfin JA, Mandiyan VS, Shah NM, Rubenstein JLR, and Mucke L(2008)Abnormal social behaviors in mice lacking Fgf17. Genes Brain Behav. 7: 344–354
deIpolyi AR, Fang S, Palop JJ, Yu G-Q, Wang X, and Mucke L (2008) Altered navigational strategy use and visuospatial deficits in hAPP transgenic mice. Neurobiol. Aging 29: 253–266
Gan L and Mucke L (2008) Paths of convergence: Sirtuins in aging and neurodegeneration. Neuron 58: 10–14
Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu G-Q, and Mucke L(2007) Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science 316: 750–754
Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu G-Q, Kreitzer A, Finkbeiner S, Noebels JL, and Mucke L(2007) Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55: 697–711
deIpolyi AR, Rankin KP, Mucke L, Miller BL, and Gorno-Tempini ML(2007) Spatial cognition and the human navigation network in AD and MCI. Neurology 69: 986–997
Chin J, Massaro CM, Palop JJ, Thwin MT, Yu G-Q, Bien-Ly N, Bender A, and Mucke L (2007) Reelin depletion in the entorhinal cortex of human amyloid precursor protein transgenic mice and humans with Alzheimer’s disease. J. Neurosci. 27: 2727–2733
Cheng IH, Scearce-Levie K, Legleiter J, Palop JJ, Gerstein H, Bien-Ly N, Puoliväli J, Lesné S, Ashe KH, Muchowski PJ, and Mucke L(2007) Accelerating Amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. JBC 282: 23818–23828
Tesseur I, Zou K, Esposito L, Bard F, Berber E, Van Can J, Lin AH, Crews L, Tremblay P, Matthews P, Mucke L, Masliah E, and Wyss-Coray T (2006) Deficiency in neuronal TGF-b signaling promotes neurodegeneration and Alzheimer’s pathology. J. Clin. Invest. 116: 3060–3069
Palop JJ, Chin J, and Mucke L (2006) A network dysfunction perspective on neurodegenerative diseases. Nature 443: 768–773
Roberson ED and Mucke L (2006) 100 years and counting: Prospects for defeating Alzheimer’s disease. Science 314: 781–784
Mueller-Steiner S, Zhou Y, Arai H, Roberson ED, Chen J, Wang X, Yu G-Q, Esposito L, Mucke L, and Gan L (2006) Anti-amyloidogenic and neuroprotective functions of cathepsin B: Implications for Alzheimer’s disease. Neuron 51: 703–714
Esposito L, Raber J, Kekonius L, Yan F, Yu GQ, Bien-Ly N, Puoliväli J, Scearce-Levie K, Masliah E, and Mucke L(2006) Reduction of mitochondrial superoxide dismutase modulates Alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J. Neurosci. 26: 5167–5179
Choi D-S, Wang D, Yu G-Q, Zhu G, Kharazia VN, Paredes JP, Chang WS, Deitchman JKMucke L, and Messing RO (2006) PKCe increases endothelin converting enzyme activity and reduces amyloid plaque pathology in transgenic mice. PNAS 103: 8215–8220
Chen J, Zhou Y, Chen L.-F, Mueller-Steiner S, Chen L-F, Kwon H, Yi S, Mucke L, and Gan L (2005) SIRT1 protects against microglia-dependent amyloid-b toxicity through inhibiting NF-kB signaling. J. Biol. Chem. 280: 40364–40374
Chin J, Palop JJ, Puoliväli J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, and Mucke L(2005)Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 25: 9694–9703
Palop JJ, Chin J, Bien-Ly N, Massaro C, Yeung BZ, Yu G-Q, and Mucke L (2005) Vulnerability of dentate granule cells to disruption of Arc expression in human amyloid precursor protein transgenic mice. J. Neurosci. 25: 9686–9693
Rockenstein E, Mante M, Alford M, Adame A, Crews L, Hashimoto M, Esposito L, Mucke L, and Masliah E (2005) High b-secretase activity elicits neurodegeneration in transgenic mice despite reductions in amyloid-b levels: Implications for the treatment of Alzheimer’s disease. JBC 280: 32957–32967
Esposito L, Gan L, Yu G-Q, Essrich C, and Mucke L (2004) Intracellularly generated amyloid-b peptide counteracts the antiapoptotic function of its precursor protein and primes proapoptotic pathways for activation by other insults in neuroblastoma cells. J. Neurochem. 91: 1260–1274
Cheng IH, Palop JJ, Esposito LA, Bien-Ly N, Yan F, Mucke L (2004) Aggressive amyloidosis in mice expressing human amyloid peptides with the Arctic mutation. Nat. Med. 10: 1190–1192
Brecht WJ, Harris FM, Chang S, Tesseur I, Yu G-Q, Xu Q, Wyss-Coray T, Buttini M, Mucke L, Mahley RW, and Huang Y (2004) Neuron-specific apolipoprotein E4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci. 24: 2527–2534
Chin J, Palop JJ, Yu G-Q, Kojima N, Masliah E, and Mucke L (2004)Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J. Neurosci. 24: 4692–4697
Harris FM, Brecht WJ, Xu Q, Tesseur I, Kekonius L, Wyss-Coray T, Fish JD, Masliah E, Hopkins PC, Scearce-Levie K, Weisgraber KH, Mucke L, Mahley RW, and Huang Y (2003) Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. PNAS. USA 100: 10966–10971
Palop JJ, Jones B, Kekonius L, Chin J, Yu G-Q, Raber J, Masliah E, and Mucke L (2003) Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. PNAS 100: 9572–9577
Buttini M, Yu G-Q, Shockley K, Huang Y, Jones B, Masliah E, Mallory M, Yeo T, Longo FM, and Mucke L (2002) Modulation of Alzheimer-like synaptic and cholinergic deficits in transgenic mice by human apolipoprotein E depends on isoform, aging and overexpression of Ab but not on plaque formation. J. Neurosci. 22: 10539–10548
Raber J, Buttini M, LeFevour A, and Mucke L (2002) Androgens protect against apoE4-induced cognitive deficits. J. Neurosci. 22: 5204–5209
Wyss-Coray T and Mucke L (2002) Inflammation in neurodegenerative disease – A double-edged sword. Neuron 35: 419–432
Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, and Mucke L(2001) b-Amyloid peptides enhance a-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. PNAS 98: 12245–12250
Wyss-Coray T, Lin C, Yan F, Yu G-Q, Rohde M, McConlogue L, Masliah E, and Mucke L (2001) TGF-b1 promotes microglial amyloid-b clearance and reduces plaque burden in transgenic mice. Nat. Med. 7: 612–618
Mucke L, Yu G-Q, McConlogue L, Rockenstein EM, Abraham CR, and Masliah E (2000) Astroglial expression of human a1-antichymotrypsin enhances Alzheimer-like pathology in amyloid protein precursor transgenic mice. Am. J. Pathol. 157: 2003–2010
Mucke L, Masliah E, Yu G-Q, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, and McConlogue L (2000) High-level neuronal expression of Ab1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 20: 4050–4058
Buttini M, Akeefe H, Lin C, Mahley RW, Pitas RE, Wyss-Coray T, and Mucke L (2000) Dominant negative effects of apolipoprotein E4 revealed in transgenic models of neurodegenerative disease. Neuroscience 97: 207–210
Raber J, Akana SF, Bhatnagar S, Dallman MF, Wong D, and Mucke L (2000) Hypothalamic-pituitary-adrenal dysfunction in Apoe-/- mice: Possible role in behavioral and metabolic alterations. J. Neurosci. 20: 2064–2071
Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, and Mucke L (2000) Dopaminergic loss and inclusion body formation in a-synuclein mice: Implications for neurodegenerative disorders. Science 287: 1265–1269
Raber J, Wong D, Yu G-Q, Buttini M, Mahley RW, Pitas RE, and Mucke L (2000) Alzheimer’s disease: Apolipoprotein E and cognitive performance. Nature 404: 352–354
Wyss-Coray T, Lin C, Sanan DA, Mucke L, and Masliah E (2000) Chronic overproduction of transforming growth factor-b1 in astrocytes promotes Alzheimer’s disease-like microvascular degeneration in transgenic mice. Am. J. Pathol. 156: 139–150
D’Hooge R, Franck F, Mucke L, and De Deyn PP (1999) Age-related behavioural deficits in transgenic mice expressing the HIV-1 coat protein gp120. Eur. J. Neurosci. 11: 4398–4402
Huang F, Buttini M, Wyss-Coray T, McConlogue L, Kodama T, Pitas RE, and Mucke L (1999) Elimination of the class A scavenger receptor does not affect amyloid plaque formation or neurodegeneration in transgenic mice expressing human amyloid protein precursors. Am. J. Pathol. 155: 1741–1747
Sofroniew MV, Bush TG, Blumauer N, Kruger L, Mucke L and Johnson MH (1999) Genetically-targeted and conditionally-regulated ablation of astroglial cells in the central, enteric and peripheral nervous systems in adult transgenic mice. Brain Res. 835: 91–95
Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, and Sofroniew MV (1999) Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 23: 1–20
Xu X, Yang D, Wyss-Coray T, Yan J, Gan L, Sun Y, and Mucke L (1999) Wild-type but not Alzheimer-mutant amyloid precursor protein confers resistance against p53-mediated apoptosis. PNAS 96: 7547–7552
Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, Mucke L, and Mahley RW (1999) Expression of human apolipoprotein E3 or E4 in the brains of Apoe-/- mice: Isoform-specific effects on neurodegeneration. J. Neurosci. 19: 4867–4880
Hsia A, Masliah E, McConlogue L, Yu G-Q, Tatsuno G, Hu K, Kholodenko D, Malenka R, Nicoll R, and Mucke L (1999) Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. PNAS 96: 3228–3233
Raber J, Wong D, Buttini M, Orth M, Bellosta S, Pitas RE, Mahley RW, and Mucke L (1998) Isoform-specific effects of human apolipoprotein E on brain function revealed in Apoe knockout mice–Increased susceptibility of females. PNAS 95: 10914–10919
Masliah E, Raber J, Alford M, Mallory M, Mattson MP, Yang D, Wong D, and Mucke L (1998) Amyloid protein precursor stimulates excitatory amino acid transport: Implications for roles in neuroprotection and pathogenesis. JBC 273: 12548–12554
Krucker T, Toggas SM, Mucke L, and Siggins GR (1998) Transgenic mice with cerebral expression of human immunodeficiency virus type-1 coat protein gp120 show divergent changes in short- and long-term potentiation in CA1 hippocampus. Neuroscience 83: 691–700
Buttini M, Westland CE, Masliah E, Yafeh AM, Wyss-Coray T, and Mucke L (1998) Novel role of human CD4 molecule identified in neurodegeneration. Nat. Med. 4: 441–446
Bush TG, Savidge TC, Freeman TC, Cox HJ, Campbell EA, Mucke L, Johnson MH, and Sofroniew MV (1998) Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell 93: 189–201
Xu X, Raber J, Yang D, Su B, and Mucke L (1997) Dynamic regulation of c-Jun N-terminal kinase activity in mouse brain by environmental stimuli. PNAS 94: 12655–12660
Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, and Mucke L (1997) Amyloidogenic role of cytokine TGF-b1 in transgenic mice and in Alzheimer’s disease. Nature 389: 603–606
Wyss-Coray T, Borrow P, Brooker MJ, and Mucke L (1997) Astroglial overproduction of TGF-b1 enhances inflammatory central nervous system disease in transgenic mice. J. Neuroimmunol. 77: 45–50
Mattson MP, Barger SW, Furukawa K, Bruce AJ, Wyss-Coray T, Mark RJ, and Mucke L (1997) Cellular signaling roles of TGFb, TNFa and bAPP in brain injury responses and Alzheimer’s disease. Brain Res. Rev. 23: 47–61
Raeber AJ, Race RE, Brandner S, Priola SA, Sailer A, Bessen RA, Mucke L, Manson J, Aguzzi A, Oldstone MBA, Weissmann C, and Chesebro B (1997) Astrocyte-specific expression of hamster prion protein (PrP) renders PrP knockout mice susceptible to hamster scrapie. EMBO J. 16: 6057–6065
Raber J, Chen S, Mucke L, and Feng L (1997) Corticotropin-releasing factor and adrenocorticotrophic hormone as potential central mediators of OB effects. J. Biol. Chem. 272: 15057–15060
Masliah E, Westland CE, Rockenstein EM, Abraham CR, Mallory M, Veinberg I, Sheldon E, and Mucke L (1997) Amyloid precursor proteins protect neurons of transgenic mice against acute and chronic excitotoxic injuries in vivo.Neuroscience 78: 135–146
Zhao J, Paganini L, Mucke L, Gordon M, Refolo L, Carman M, Sinha S, Oltersdorf T, Lieberburg I, and McConlogue L (1996) b-secretase processing of the b-amyloid precursor protein in transgenic mice is efficient in neurons but inefficient in astrocytes. J. Biol. Chem. 271: 31407–31411
Wyss-Coray T, Masliah E, Toggas SM, Rockenstein EM, Brooker MJ, Lee HS, and Mucke L (1996) Dysregulation of signal transduction pathways as a potential mechanism of nervous system alterations in HIV-1 gp120 transgenic mice and humans with HIV-1 encephalitis. J. Clin. Invest. 97: 789–798
Toggas SM, Masliah E, and Mucke L (1996) Prevention of HIV-1 gp120-induced neuronal damage in the central nervous system of transgenic mice by the NMDA receptor antagonist memantine. Brain Res. 706: 303–307
Raber J, Toggas SM, Lee S, Bloom FE, Epstein CJ, and Mucke L (1996) Central nervous system expression of HIV-1 gp120 activates the hypothalamic-pituitary-adrenal axis: Evidence for an involvement of NMDA receptors and nitric oxide synthase. Virology 226: 362–373
Mohajeri MH, Bartsch U, van der Putten H, Sansig G, Mucke L, and Schachner M (1996) Neurite outgrowth on non-permissive substrates in vitro is enhanced by ectopic expression of the neural adhesion molecule L1 by mouse astrocytes. Eur. J. Neurosci. 8: 1085–1097
Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, and Games D (1996) Comparison of the neurodegenerative pathology in transgenic mice overexpressing V717F b-amyloid precursor protein and Alzheimer’s disease. J. Neurosci. 16: 5795–5811
D’Hooge R, Nagels G, Westland CE, Mucke L, and De Deyn PP (1996) Spatial learning deficit in mice expressing human 751-amino acid b-amyloid precursor protein. Neuroreport 7: 2807–2811
Wyss-Coray T, Feng L, Masliah E, Ruppe MD, Lee HS, Toggas SM, Rockenstein EM, and Mucke L (1995) Increased central nervous system production of extracellular matrix components and development of hydrocephalus in transgenic mice overexpressing transforming growth factor-b1. Am. J. Pathol. 147: 53–67
Verderber L, Johnson W, Mucke L, and Sarthy V (1995) Differential regulation of a GFAP-lacZ transgene in retinal astrocytes and Müller cells. IOVS. 36: 1137–1143
Rockenstein E, McConlogue L, Tan H, Gordon M, Power M, Masliah E, and Mucke L (1995) Levels and alternative splicing of amyloid b protein precursor (APP) transcripts in brains of APP transgenic mice and humans with Alzheimer’s disease. J. Biol. Chem. 270: 28257–28267
Rall GF, Mucke L, and Oldstone MBA (1995) Consequences of cytotoxic T lymphocyte interaction with MHC class I-expressing neurons in vivo. J. Exp. Med. 182: 1201–1212
Race RE, Priola SA, Bessen RA, Ernst D, Dockter J, Rall GF, Mucke L, Chesebro B, and Oldstone MBA (1995) Neuron-specific expression of a hamster prion protein minigene in transgenic mice induces susceptibility to hamster scrapie agent. Neuron 15: 1183–1191
Mucke L, Abraham CR, Ruppe MD, Rockenstein EM, Toggas SM, Alford M, and Masliah E (1995) Protection against HIV-1 gp120-induced brain damage by neuronal expression of amyloid precursor protein. J. Exp. Med. 181: 1551–1556
Johnson WB, Ruppe MD, Rockenstein EM, Price J, Sarthy VP, Verderber LC, and Mucke L (1995) Indicator expression directed by regulatory sequences of the glial fibrillary acidic protein (GFAP) gene: In vivo comparison of distinct GFAP-lacZ transgenes. Glia 13: 174–184
Games D, Adams D, Alessandrini R, Barbour R, Borthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala M, Mucke L, Paganini L, and Penniman L (1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717F b-amyloid precursor protein. Nature 373: 523–527
Borrow P, Cornell JL, Ruppe MD, and Mucke L (1995) Immunization-induced inflammatory infiltration of the central nervous system in transgenic mice expressing a foreign antigen in astrocytes. J. Neuroimmunol. 61: 133–149
Toggas SM, Masliah E, Rockenstein EM, Rail GF, Abraham CR, and Mucke L (1994) Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature 367: 188–193
Rall GF, Mucke L, Nerenberg M, and Oldstone MBA (1994) A transgenic mouse model to assess the interaction of cytotoxic T lymphocytes with virally infected, class I MHC-expressing astrocytes. J. Neuroimmunol. 52: 61–68
Mucke L, Masliah E, Johnson WB, Ruppe MD, Alford M, Rockenstein EM, Forss-Petter S, Pietropaolo M, Mallory M, and Abraham CR (1994) Synaptotrophic effects of human amyloid b protein precursor in the cortex of transgenic mice. Brain Res. 666: 151–167
Mucke L and Rockenstein EM (1993) Prolonged delivery of transgene products to specific brain regions by migratory astrocyte grafts. Transgenics 1: 3–9
Campbell IL, Abraham CR, Masliah E, Kemper P, Inglis JD, Oldstone MBA, and Mucke L (1993) Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. PNAS 90: 10061–10065
Eddleston M and Mucke L (1993) Molecular profile of reactive astrocytes–Implications for their role in neurologic disease. Neuroscience 54: 15–36
Mucke L and Eddleston M (1993) Astrocytes in infectious and immune-mediated diseases of the central nervous system. FASEB J. 7: 1226–1232
Abraham CR, Kanemaru K, and Mucke L (1993) Expression of cathepsin G-like and a1-antichymotrypsin-like proteins in reactive astrocytes. Brain Res. 621: 222–232
Mucke L and Oldstone MBA (1992) The expression of major histocompatibility complex (MHC) class I antigens in the brain differs markedly in acute and persistent infections with lymphocytic choriomeningitis virus (LCMV). J. Neuroimmunol. 36: 193–198
Mucke L, Oldstone MBA, Morris JC, and Nerenberg MI (1991) Rapid activation of astrocyte-specific expression of GFAP-lacZ transgene by focal injury. New Biol. 3: 465–474
Joly E, Mucke L, and Oldstone MBA (1991) Viral persistence in neurons explained by a lack of major histocompatibility class I expression. Science 253: 1283–1285
Ross ME, Evinger MJ, Hyman SE, Carroll JM, Mucke L, Comb M, Reis DJ, Joh TH, and Goodman HM (1990) Identification of a functional glucocorticoid response element in the phenylethanolamine N-methyltransferase promoter using fusion genes introduced into chromaffin cells in primary culture. J. Neurosci. 10: 520–530
Berger JR and Mucke L (1988) Prolonged survival and partial recovery in AIDS-associated progressive multifocal leukoencephalopathy. Neurology 38: 1060–1065
Schalling M, Dagerlind A, Breve S, Petterson R, Kvist S, Brownstein M, Hyman SE, Mucke L, Goodman HM, Joh TH, Goldstein M, and Hökfelt T (1987) Localization of mRNA for phenylethanolamine N-methyltransferase (PNMT) using in situ hybridization. Acta Physiol. Scand. 131: 631–632
Norita M, Mucke L, Benedek G, Albowitz B, Katoh Y, and Creutzfeldt OD (1986) Connections of the anterior ectosylvian visual area (AEV). Exp. Brain. Res. 62: 225–240
Mucke L, Norita M, Benedek G, and Creutzfeldt OD (1982) Physiologic and anatomic investigation of a visual cortical area situated in the ventral bank of the anterior ectosylvian sulcus of the cat. Exp. Brain. Res. 46: 1–11